Exploring mechanisms for Omega-3 anti-colorectal cancer effect

By Alex Davie
Omega-3 fatty acids (O3FAs) are known to have an anti-colorectal cancer (CRC) effect, although the mechanism(s) behind this anti-CRC activity are unclear. One potential mechanism is the increased production of short-chain fatty acids (SCFAs) by resident gut bacteria in response to O3FA concentration.
Project overview
Through this project we aimed to explore one of the potential mechanisms for O3FAs anti-CRC effect: the enhanced production of SCFAs by resident colorectal bacteria. Having been previously demonstrated that O3FA treatment leads to an increase in abundance of bacteria which are believed to be involved in SCFA production we aimed to test whether O3FA presence increases luminal SCFA levels in an in vitro model. O3FA is believed to be probiotic and if so fibre could be the prebiotic that, when present in sufficient quantities, works with O3FAs to augment the production of SCFAs. The findings of this study, combined with further research, will help to inform future clinical studies aiming to use O3FAs in combination with an increased dietary fibre intake for prevention and/or treatment of bowel cancer. In addition to this we also had the aim of performing a comparative analysis of different metagenomics analytical tools to examine the pros and cons of each.
Data and methods
In this study the DNA and RNA sequencing data was extracted from in vitro models produced using gut microbiome extractions from a total of 6 different volunteers. The DNA extraction data was produced from 6 different volunteers each with 5 different experimental conditions/time points, totalling 60 experimental samples as each sample had both a forward and reverse read. Similarly, the RNA extraction data was produced from 3 different volunteers each with 9 different experimental conditions/time points, totalling 54 experimental samples due to forward and reverse reads. The experimental conditions used were control, control and inulin, O3FA and O3FA and inulin. Extractions were taken from each of these conditions at different timepoints to measure any change in microbial species abundance and/or activity in response to fibre and/or O3FAs.
The data in this study was generated through the use of shotgun metagenomic sequencing, allowing all of the species within each of the in vitro models to be sequenced in parallel. The output of this shotgun metagenomic sequencing was DNA/RNA sequence reads which then needed to be processed with metagenomic analysis tools in order to obtain interpretable outputs. The metagenomic analysis tools used in this project were DIAMOND, MEGAN and MetaPhlAn4. The raw reads were stored and processed using Arc3 due to the size of the files and computational resources required for the analysis. The first step of the analysis was taxonomic and functional binning of the microbiome sequences, this is the grouping and identification sequences within the samples based on a reference database. For DIAMOND/MEGAN this was the NCBI protein database, whereas MetaPhlAn uses a comprehensive set of 1.3 million unique marker genes to identify the microorganisms present in the samples.
After aligning the DNA/RNA sequences to the databases the next step was to analyse the results of the aligning step. In the case of DIAMOND/MEGAN this step allowed the analysis of species present as well as providing functional insights, i.e. the presence of specific pathways and the production of principle component analyses as well as many other analysis techniques. MetaPhlAn on the other hand provided a list of relative abundances for species present within the samples, allowing the identification of species with higher abundance in the presence of O3FA/inulin as well as the production of heatmaps showing which species are more functionally active.
Key findings
This project is part of a greater ongoing study however it has still provided numerous insights. First it has revealed the benefits of using two metagenomic analysis tools. DIAMOND/MEGAN are both very resource heavy, both in terms of time and computation power. However, it is much more user friendly as the analysis of results is GUI based and more readily provides different methods of analysis once the sequences have been aligned with the reference database. Comparatively, MetaPhlAn is much quicker and requires far fewer computation resources – producing relative abundance tables in a matter of minutes compared to the hours required by DIAMOND/MEGAN. However, beyond the production of relative abundance tables and comparative heatmaps carrying out any further analysis is less straightforward and requires the use of additional metagenomic tools.
Using the relative abundance tables produced by MetaPhlAn the first step was to identify which taxonomic families were the most present. To do this heatmaps were produced, plotting the relative abundance of taxonomic families within each sample alongside each other. This revealed a few taxonomic families that were significantly more abundant than others: Prevotellaceae, Bifidobacteriaceae and Bacteriaceae were all taxonomic families identified with a higher abundance in samples containing inulin and O3FAs, shown in figure 1. These results will lead onto the next stage of the research as the focus narrows to identify particular taxonomic genus’s known to be linked to SCFA production within these taxonomic families.
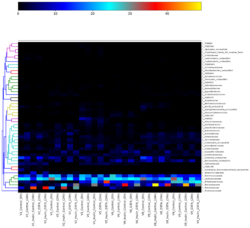
Figure 1. Heatmap of Top 50 DNA Taxonomic Families. This heatmap displays the relative abundances of the top 50 bacterial species. The heatmap uses colour gradients to represent data, with each row indicating a different taxonomic species and each column representing one of the volunteers’ samples. Colours range from cool to warm, signifying low to high abundance. The taxonomic families of interest are all located towards the bottom of the heatmap with the most interesting families being Prevotellaceae, Bacteriaceae and Bifidobacteriaceae. A clear legend is provided at the top of the plot to assist with identifying relative abundances.
Analysis of the data produced with DIAMOND/MEGAN allowed the identification of pathways of interest as well as the production of Principal Coordinate Analysis (PCoA) plots. These pathways of interest were identified using the Kyoto Encyclopaedia of Genes and Genomes. These KEGG pathways are maps representing networks of interacting molecules and genes responsible for specific cellular functions. Further analysis of these pathways will enable the identification of SCFA producing species, as well as identifying any other pathways they are involved in. Through DIAMOND/MEGAN it was also possible to produce PCoA plots of the different experimental conditions, for example the different DNA samples from volunteer 1. This allowed us to visualize how the different samples cluster together based on the microbiota present, and in some cases revealed extra families that hadn’t been previously identified having a large effect on the O3FA/Inulin samples, for example Fermicuties and Terrabacteria group (figure 2).

Figure 2. Principal Coordinate Analysis for Volunteer 1. This plot shows a Principal Coordinate Analysis (PCoA) plot generated in MEGAN, illustrating the relationships among nine distinct metagenomic samples. The plot uses coordinates to represent the multidimensional data in a two-dimensional space, allowing for the visualization of sample similarities and differences. Points on the plot represent the samples, and their positions are determined by their similarity in the high-dimensional data. This PCoA plot aids in understanding the overall structure of the metagenomic dataset, as well as identifying which taxonomic families are having the largest effect on the different samples.
Value of the research
What is the impact, how will this benefit xyz? How could the research be applied to real-world problems or what is the usage/benefit?
This project was part of ongoing research into the benefits of O3FAs on the production of SCFAs. This project is a first of its kind, exploring the potential of a novel transdisciplinary (science, technology, medicine, bioinformatics, statistics, and chemistry) intervention with the result informing future clinical studies. In addition this project proved that DNA and RNA samples can be cultivated and collected from the same plate, whilst still producing high quality samples. These future clinical studies will hopefully improve the use of O3FAs for prevention and/or treatment of bowel cancer. The current aim is to publish this research with the hopeful end goal of providing evidence that supplementing the diet with O3FAs can be used alongside traditional therapeutics for the treatment of colorectal cancer.
Insights
- MetaPhlAn4 enables much quicker analysis than DIAMOND/MEGAN, however the outputs are more limited and does not enable the use of the KEGG pathway.
- Furthered understanding of the effects of O3FAs and inulin on the production of SCFAs, particularly genus’s, pathways, and enzymes more abundant/active as a response to increase O3FA.
- Research will continue beyond the end of this project with more focus on results of interest.
Research theme
Identify which LIDA research theme(s) this sits under:
- Health
Programme theme (select all that apply)
- Statistical Data Science
- Mathematical and Computational Foundations
People
Alexander Davie – Data Scientist, Leeds Institute for Data Analytics
Dr Suparna Mitra – University Academic Fellow in Gastrointestinal Research including Bioinformatics, University of Leeds
Professor Mark Hull – Professor of Molecular Gastroenterology, University of Leeds
Joanna Aldoori - PhD
Partners
NA
Funders
Leeds Crucible Funding for Metagenomic Sequencing